Image posted with permission from J. Chem. Theory Comput., 2013, 9 (11), pp 5004–5020. Copyright 2013 American Chemical Society
In a recently published paper in the Journal of Chemical Theory
and Computation of the American Chemical Society (ACS), density
functional theory (DFT) calculations were found to correctly reproduce
the 57Fe isomer shifts (δ) and quadrupole splittings (ΔEQ)
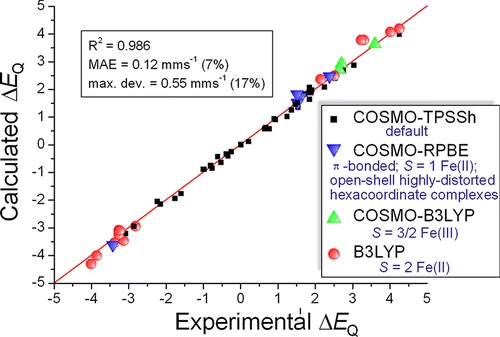
of a large and chemically very diverse set of 66 Fe complexes. For the
isomer shifts, several density functionals provide accurate δ values for
all investigated iron compounds, with the best-performing DFT method
yielding a mean absolute error (MAE) of 0.05 mms-1 and a
maximum deviation of 0.12 mms-1. Although a similarly
well-performing functional could not be chosen for the prediction of
quadrupole splittings, the selection of an appropriate DFT method by a
careful chemical classification of Fe complexes enables the accurate
prediction of this parameter: the application of this approach yields a
MAE of 0.12 mms-1 (7% error) and a maximum deviation of 0.55
mms-1 (17% error) (see the figure presented above). This
accuracy should be sufficient for most chemical problems that concern Fe
complexes. Besides these benchmark results, special issues are also
covered in the article including the prediction of ΔEQ in
especially challenging cases, quadrupole splittings at phase transitions
induced by variations of the electronic structure (e.g. spin crossover
and inversion of the orbital ground state), as well as the reliable
determination of the sign of the quadrupole splitting. The excellent
agreement observed between the experimental and calculated signs of ΔEQ
may immensely enhance the potential of the application of Mössbauer
spectroscopy in structural research. The details of the summarized work
is available at the link below.
Link to the article:
http://pubs.acs.org/doi/abs/10.1021/ct4007585